Abstract
The secondary structure of commonly sequenced
mitochondrial and nuclear rRNA-encoding genes can be informative during
both the process of multiple-sequence alignment and the subsequent
phylogenetic analysis of these alignments. Use of secondary
structure in these cases is analogous in many ways to the definition of
morphological characters as commonly practiced in most revisionary
cladistic works. Parallels between traditional morphological
characters and characters based on secondary structure of rRNA
molecules are examined. Example alignments, and the tools to
parse, characterize, and analyze these alignments are provided.
 Molecular-based
phylogenies are now routinely used to make revisionary-systematic
decisions both under the traditional Linnean nomenclatural system and
more recently proposed systems such as the Phylocode. Character data supporting these phylogenies is usually derived from
nucleotide (DNA/RNA) or amino acid (protein) sequences and often
combined with morphological, behavioral, etc. data. Recently,
important steps have been made towards characterizing the secondary
structure of popularly used rRNA molecules, such as the 28S D2 and D3
expansion segments (Gillespie et al., submitted: see greyed area of Fig. 1) and the
variable region 4 of the 18S rRNA (Wuyts et al., 2000). These
predicted structures of the more variable regions of rRNA are
extensions of over a decade of refinements to structure predictions for
the mitochondrial 16S and 12S genes and nuclear 28S, 18S, 5.8S and 5S
genes (e.g., Cannone et al. 2002). In a phylogenetic context,
these characterizations may be seen as a hybrid between DNA and
morphological characters. In practice, both traditional single
column (nucleotide) characters and multi-nucleotide structures defined
as single states may be derived from multiple-sequence alignments that
are based on secondary structure models.
Molecular-based
phylogenies are now routinely used to make revisionary-systematic
decisions both under the traditional Linnean nomenclatural system and
more recently proposed systems such as the Phylocode. Character data supporting these phylogenies is usually derived from
nucleotide (DNA/RNA) or amino acid (protein) sequences and often
combined with morphological, behavioral, etc. data. Recently,
important steps have been made towards characterizing the secondary
structure of popularly used rRNA molecules, such as the 28S D2 and D3
expansion segments (Gillespie et al., submitted: see greyed area of Fig. 1) and the
variable region 4 of the 18S rRNA (Wuyts et al., 2000). These
predicted structures of the more variable regions of rRNA are
extensions of over a decade of refinements to structure predictions for
the mitochondrial 16S and 12S genes and nuclear 28S, 18S, 5.8S and 5S
genes (e.g., Cannone et al. 2002). In a phylogenetic context,
these characterizations may be seen as a hybrid between DNA and
morphological characters. In practice, both traditional single
column (nucleotide) characters and multi-nucleotide structures defined
as single states may be derived from multiple-sequence alignments that
are based on secondary structure models. | 1. Figure 2 illustrates (in
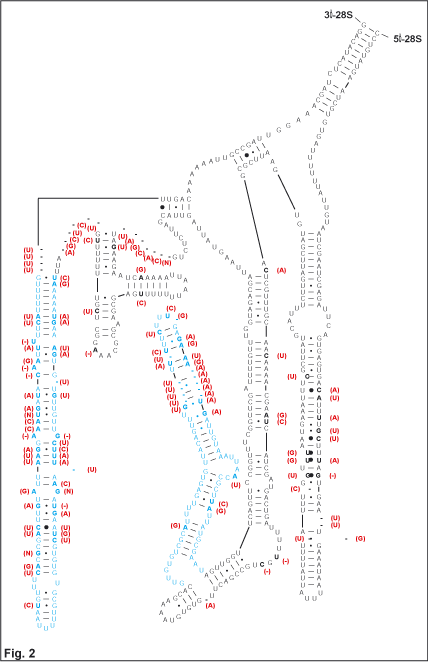
blue) several major modifications to the expansion segment D2 of the
28S rRNA for myrmecolacid Strepsiptera. Red bases represent
substitutions between populations of Caenocholax fenyesi from Texas and Mexico. Gross modifications on this scale are to-date uncommonly found. |
|
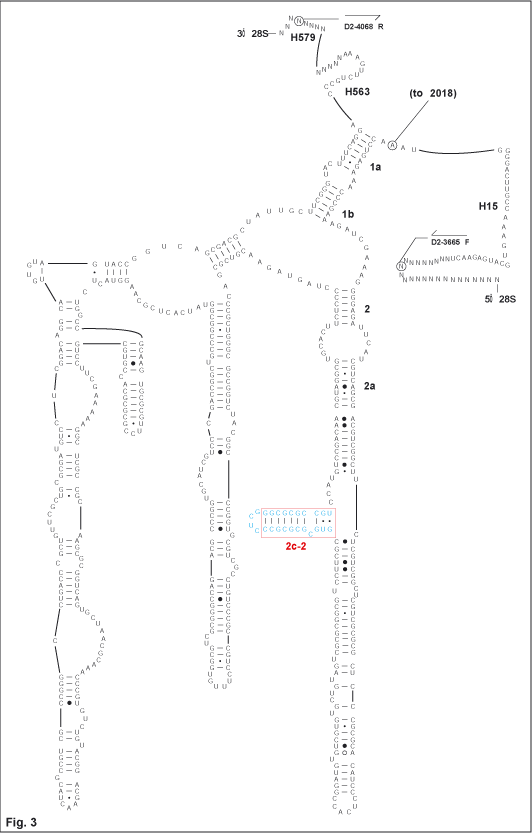
| 2. Figure 3 illustrates a novel helix, 2c-2,
apparently synapomorphic for a clade of basal evaniids. Though
phylogenetic analyses are ongoing, preliminary evidence suggests that
this character supports a putatively monophyletic clade further supported by wing
venation characters (Deans, pers. com.). This structure is not
known in other insects. |
 |
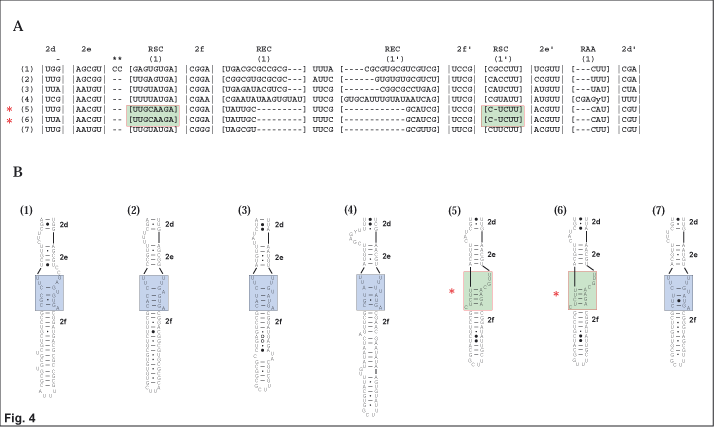
| 3. Figure 4 illustrates a region of slipped-strand compensation that results in a novel structure for a clade of Acalymma spp. sensu stricto (striped cucumber beetles). Here regions of slipped-strand compensation bounded by helices 2e and 2f form in two unique ways (represented by blue and green boxes). A deletion in RSC (1’) in Acalymma results in a distinct structure not found in any other sampled diabroticine beetles (Gillespie et al. 2004b). |
 |
| 4. For a fourth alignment pertaining to ichneumonoids (Gillespie, Yoder, Wharton, submitted) characters derived from differences in compostion and length for RAAs, RSCs, and RECs (bracketed blocks in the alignment) were analyzed under a parsimony framework in POY and PAUP* following recoding in INAASE (results not shown). Partial results of these analyses are found in figure 10b (in particular note white squares, which represent branches supported when the content of each block for each taxon was treated as a single character). While topologies derived from these characters were generally less strongly supported (bootstraps, consensus topologies) these characters were found to support numerous clades recovered using non-secondary structure based alignments. |