phylogenetic analyses
Below a small (and by no means comprehensive) list of ways to incorporate information pertaining to secondary structure contained in the alignments into working phylogenetic analyses.
MrBayes
All modeling as seen in the Likelihood and Modeltest sections below is available in Bayesian analyses. In addition, the jRNA scripts automatically generate pairs statements for using in the doublet model available in Mr. Bayes. "Databreaks" statements area also generated for each block of data exported, allowing this functionality in Mr. Bayes to be used.
PHASE 1.1
All modeling as in the Likelihood and Modeltest sections below is available in analyses in PHASE 1.1. The jRNA scripts generate PHASE legal input files and command files regardless of the complexity of the bracketing (helical regions) in the mask. Pseudoknot coding is thus supported. PHASE 2.0 will be supported in the 'Psy' package.
POY
Alignments based on secondary structure are fully compatible in POY. In fact, they can be seen as strictly adhering to the recommendations for parsing data prior to using POY. Two major advantages are inherently available in the alignments described herein: 1) the average block length is very short, and in theory much easier to optimize in POY (see POY manual); and 2) all combinations of fixed-state and direct optimization on aligned and unaligned blocks are possible. For 2), analyses A-H in the table are possible. These combinations are easily generated using the jRNA scripts.
| Fixed states |
Direct optimization |
|
| Aligned alone |
A G |
C H |
| Unaligned alone |
B H |
D G |
| All data |
E |
F |
Parsimony
Parsimony analyses are generally only run on aligned (unbracketed) regions of the matrix. The conversion to fixed-states characters of unaligned regions, using programs such as INAASE, are also possible.
Likelihood
Separate models can be derived for helical and non-helical regions, or individual blocks.
Modeltest
The jRNA scripts allow for export of helical, or non-helical regions to be exported as separate matrices. These then can be run through Modeltest to generate separate models of evolution for both types of data. In theory each block (space delimited) could be modeled individually.
Jrna.pm matrix format
Jrna.pm requires two files. A Nexus legal data matrix and a block index file. Elements in each file that are required or unique to the format are highlighted in color.
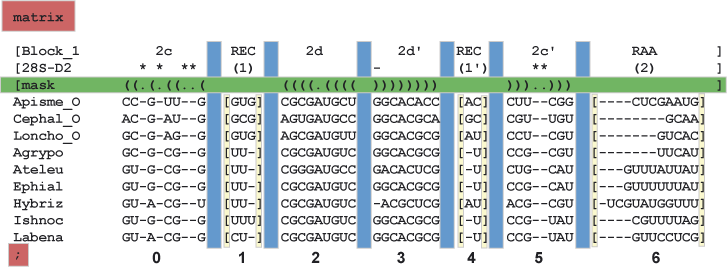
- Red - a line with "matrix" before the matrix, and a closing semi-colon.
- Green - a PHASE style mask, with "[mask" at the line beginning and "]" at the line end.
- Blue - white-space (spaces only, not tabs) between blocks.
- Yellow - brackets surround unaligned blocks (e.g. 1, 4, 6). These blocks are included in some analyses and should not be removed! These regions allow for the full sequence to be presented in each alignment.
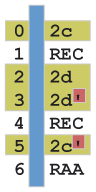
- Blue - white-space (spaces or tabs) between the two columns.
- Yellow - represent blocks that are defined as helices in this alignment.
- Red - helices are identified by the presence of two names (e.g. "2c", "2c'") differing only by an apostrophe.